Traditional, non-spatial models
Source:vignettes/vignette-04-nonspatial-models.Rmd
vignette-04-nonspatial-models.RmdThe biggest selling point of the slendr package is that you can program spatiotemporal population genetics models in R and have them execute automatically in SLiM. However, there are several reasons why you might be interested in using slendr even for non-spatial models. First, R is a language that many scientists already know, and being able to simulate data from the comfort of an R interface significantly lowers the barrier of entry. Second, because slendr makes SLiM appear almost as if it were just another R library, running simulations (spatial and non-spatial), fitting models, exploring parameter grids, calculating statistics, and visualization of results can be all be performed without leaving the R interface.
In this vignette, we will demonstrate how to program non-spatial
models in slendr. However, we should start by noting that there
is almost no difference between code for non-spatial and spatial models
in slendr. The only visible difference is that spatial models
include a map = argument in the population()
constructor function of ancestral population(s), and non-spatial models
do not. That’s it, that’s the difference. Switching between spatial and
non-spatial models is performed internally by the package, without any
user intervention.
To make the comparison clearer, we will use the example from the slendr landing page, but we will implement it in a non-spatial context (i.e., as a traditional random mating simulation).
First, let’s define population objects, splits, and other demographic
events (note the missing map argument, which is set to
FALSE by default):
#> The interface to all required Python modules has been activated.
# African ancestral population
afr <- population("AFR", time = 100000, N = 3000)
# first migrants out of Africa
ooa <- population("OOA", parent = afr, time = 60000, N = 500, remove = 23000) %>%
resize(N = 2000, time = 40000, how = "step")
# Eastern hunter-gatherers
ehg <- population("EHG", parent = ooa, time = 28000, N = 1000, remove = 6000)
# European population
eur <- population("EUR", parent = ehg, time = 25000, N = 2000) %>%
resize(N = 10000, how = "exponential", time = 5000, end = 0)
# Anatolian farmers
ana <- population("ANA", time = 28000, N = 3000, parent = ooa, remove = 4000)
# Yamnaya steppe population
yam <- population("YAM", time = 7000, N = 500, parent = ehg, remove = 2500)We can define gene flow events in the same way as we did for the spatial model:
gf <- list(
gene_flow(from = ana, to = yam, rate = 0.5, start = 6500, end = 6400),
gene_flow(from = ana, to = eur, rate = 0.5, start = 8000, end = 6000),
gene_flow(from = yam, to = eur, rate = 0.75, start = 4000, end = 3000)
)The compilation step is also the same. The only (internal) difference is that we skip the rasterization of vector maps that is performed for spatial models in order to control and restrict population boundaries:
model <- compile_model(
populations = list(afr, ooa, ehg, eur, ana, yam),
gene_flow = gf, generation_time = 30,
time_units = "years before present"
)Using the plot_map() function doesn’t make sense, as
there are no spatial maps to plot. However, we can still plot the
demographic graph, verifying that the model has been specified correctly
using the function plot_model() as shown below.
Let’s say we also want to schedule specific sampling events at
specific times (which record only specified individuals in a tree
sequence). We can use schedule_sampling() to do just
that:
samples <- schedule_sampling(
model,
times = c(0, 5000, 12000, 20000, 35000, 39000, 43000),
list(eur, 3), list(ehg, 1), list(yam, 1), list(ana, 3), list(ooa, 1), list(afr, 1)
)Because it’s not just the model itself that’s useful to visually
verify (which is the main purpose of plot_model()) but also
the sampling scheme, slendr makes it possible to overlay the
numbers of individuals scheduled for tree-sequence recording from each
lineage at each timepoint:
plot_model(model, samples = samples)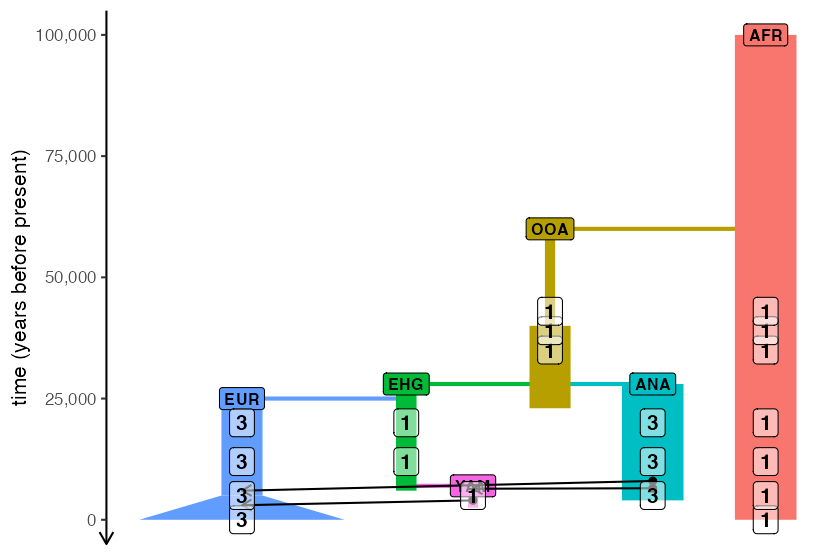
Even the final step—execution of the model in SLiM—is the same, using
the built-in slim() function:
ts_slim <- slim(model, sequence_length = 100000, recombination_rate = 0)Even for non-spatial models, this function still uses the same SLiM back end script used for spatial models. The only difference is that all spatial features are switched off, making the model run as a simple random-mating simulation.
Given that we are running a non-spatial simulation, you might wonder if it wouldn’t be more efficient to use a coalescent simulator. Indeed, slendr also provides an alternative msprime back end just for this purpose. We could run the exact same simulation with msprime like this:
ts_msprime <- msprime(model, sequence_length = 100000, recombination_rate = 0)In fact, because both SLiM and msprime back ends save outputs in a tree sequence format, we can analyse them using the same tools. See this vignette for more information about tree sequence analysis with slendr, and for more discussion on alternative simulation back ends and more extensive examples of data analysis with tree sequences you can read this tutorial.
Extracting parameters from a model or tree sequences
In some situations (such as when model parameters are drawn from
random distributions and we need to know which parameters had been used
after the simulations), function
extract_parameters() can be used. This function peeks into
a slendr tree sequence object and extract parameters of the
original slendr model:
extract_parameters(ts_msprime)#> $splits
#> pop parent N time remove
#> 1 AFR <NA> 3000 100000 NA
#> 2 OOA AFR 500 60000 23000
#> 3 EHG OOA 1000 28000 6000
#> 5 ANA OOA 3000 28000 4000
#> 4 EUR EHG 2000 25000 NA
#> 6 YAM EHG 500 7000 2500
#>
#> $gene_flows
#> from to start end rate
#> 1 ANA YAM 6500 6400 0.50
#> 2 ANA EUR 8000 6000 0.50
#> 3 YAM EUR 4000 3000 0.75
#>
#> $resizes
#> pop how N time end
#> 1 OOA step 2000 40000 NA
#> 2 EUR exponential 10000 5000 0For completeness, although this isn’t relevant in this example either, the function can also get parameters from any compiled model. For instance, we can check which parameters were used to compile the built-in slendr introgression model by running:
introgression_model <- read_model(path = system.file("extdata/models/introgression", package = "slendr"))
extract_parameters(introgression_model)#> $splits
#> pop parent N time remove
#> 1 CH <NA> 10 6500000 NA
#> 2 AFR CH 10 6000000 NA
#> 3 NEA AFR 10 600000 40000
#> 4 EUR AFR 5000 70000 NA
#>
#> $gene_flows
#> from to start end rate
#> 1 NEA EUR 55000 45000 0.03As we can see, extract_parameters() returns a list of
data frames, one data frame for each aspect of a demographic model
(where applicable).
Named samples
As an addendum, it is worth mentioning that in addition to
automatically naming recorded samples according to the format of
"<population>_<number>", slendr
supports uniquely named samples. For instance, imagine that we want to
record an ancient individual representing a 45.000 years old hunter
gatherer known as Ust’-Ishim (Fu, _et al., Nature, 2014). We could
include this sample among our other, generically named, samples like
this:
schedule_amh <- schedule_sampling(
model,
times = c(0, 5000, 12000, 20000, 35000, 39000, 43000),
list(eur, 3), list(ehg, 1), list(yam, 1), list(ana, 3), list(ooa, 1), list(afr, 1)
)
schedule_ui <- schedule_sampling(model, times = 45000, list(ooa, 1, "Ust_Ishim"))
# bind the two tables together into a single schedule
schedule <- rbind(schedule_amh, schedule_ui)Then we would simulate just like before:
ts <- msprime(model, sequence_length = 100000, recombination_rate = 0, samples = schedule)When we inspect the table of all recorded individuals, we can see that the Ust’-Ishim individual is stored under its proper symbolic name:
ts_samples(ts)#> name time pop
#> 1 Ust_Ishim 45000 OOA
#> 2 AFR_1 43000 AFR
#> 3 OOA_1 43000 OOA
#> 4 AFR_2 39000 AFR
#> 5 OOA_2 39000 OOA
#> 6 AFR_3 35000 AFR
#> 7 OOA_3 35000 OOA
#> 8 AFR_4 20000 AFR
#> 9 ANA_1 20000 ANA
#> 10 ANA_2 20000 ANA
#> 11 ANA_3 20000 ANA
#> 12 EHG_1 20000 EHG
#> 13 EUR_1 20000 EUR
#> 14 EUR_2 20000 EUR
#> 15 EUR_3 20000 EUR
#> 16 AFR_5 12000 AFR
#> 17 ANA_4 12000 ANA
#> 18 ANA_5 12000 ANA
#> 19 ANA_6 12000 ANA
#> 20 EHG_2 12000 EHG
#> 21 EUR_4 12000 EUR
#> 22 EUR_5 12000 EUR
#> 23 EUR_6 12000 EUR
#> 24 AFR_6 5000 AFR
#> 25 ANA_7 5000 ANA
#> 26 ANA_8 5000 ANA
#> 27 ANA_9 5000 ANA
#> 28 EUR_7 5000 EUR
#> 29 EUR_8 5000 EUR
#> 30 EUR_9 5000 EUR
#> 31 YAM_1 5000 YAM
#> 32 AFR_7 0 AFR
#> 33 EUR_10 0 EUR
#> 34 EUR_11 0 EUR
#> 35 EUR_12 0 EURWhen we later compute summary statistics on tree sequences, this makes referring to specific individuals even more convenient:
ts_f3(ts, A = "AFR_1", B = "EUR_1", C = "Ust_Ishim", mode = "branch")#> # A tibble: 1 × 4
#> A B C f3
#> <chr> <chr> <chr> <dbl>
#> 1 AFR_1 EUR_1 Ust_Ishim 1210.